Analysis of single-cell ATAC-seq
data using ChrAccR
data using ChrAccR
Fabian Mueller
2023-03-17
singlecell.Rmd
Introduction
Single-cell ATAC datasets can be analyzed much like bulk datasets using ChrAccR. Please refer to the main vignette for a guide on how to analyze bulk data. First, let us load the package and prepare the plot style used in the remainder of this vignette.
Loading the example data
In addition to bulk data, the ChrAccRex data package contains a small example single-cell ATAC-seq datasets. If you haven’t already installed ChrAccRex use
devtools::install_github("GreenleafLab/ChrAccRex")The example data used in this vignette was designed for illustrating the functionality of ChrAccR rather than a full analysis. It contains ATAC-seq data for a subset of 3000 cells derived from peripheral blood and bone marrow samples describing human hematopoiesis (Granja et al. 2019) and it focuses on only a subset of the human genome (chromosomes 1 and 21). It can be loaded using:
dsa <- ChrAccRex::loadExample("dsAtacSc_hema_example")
DsATACsc data structure
Analysis of single-cell datasets in ChrAccR works much like bulk datasets. ChrAccR implements a special subclass of DsATAC for storing and operating on single-cell datasets. This subclass is called DsATACsc. The main difference is that the ‘’samples’’ are actually individual cells:
dsa> Single-cell DsATAC chromatin accessibility dataset
> [in memory object]
> contains:
> * 3000 samples: BMMC_D5T1_AAACTCGCAAATTGAG-1, BMMC_D5T1_AAACTGCCACCAAGGA-1, BMMC_D5T1_AAACTGCCAGTAAGAT-1, BMMC_D5T1_AAACTGCCAGTCCTGG-1, BMMC_D5T1_AAACTGCCATTACTTC-1, ...
> * fragment data for 3000 samples
> * 3 region types: tiling5kb, promoter, .peaks.itlsi
> * * 59477 regions of type tiling5kb
> * * 2318 regions of type promoter
> * * 56045 regions of type .peaks.itlsiQuality control statistics
The annotation table stored with the dataset object can contain a number of statistics that are useful for cell quality control and filtering.
cell_anno <- getSampleAnnot(dsa)
colnames(cell_anno)> [1] ".sampleId" "cellId"
> [3] "cellBarcode" "sampleId"
> [5] "cellType" "platform"
> [7] "geoId" "fragmentFile"
> [9] "label_granja" "nFrags"
> [11] ".tssEnrichment_unsmoothed" ".tssEnrichment"
cutoff_tss <- 15
cutoff_nfrags <- 4000
ggplot(cell_anno) + aes(x=log10(nFrags), y=.tssEnrichment) +
geom_hline(yintercept=cutoff_tss, color="green") +
geom_vline(xintercept=log10(cutoff_nfrags), color="green") +
geom_point(size=0.5) + facet_wrap(~sampleId)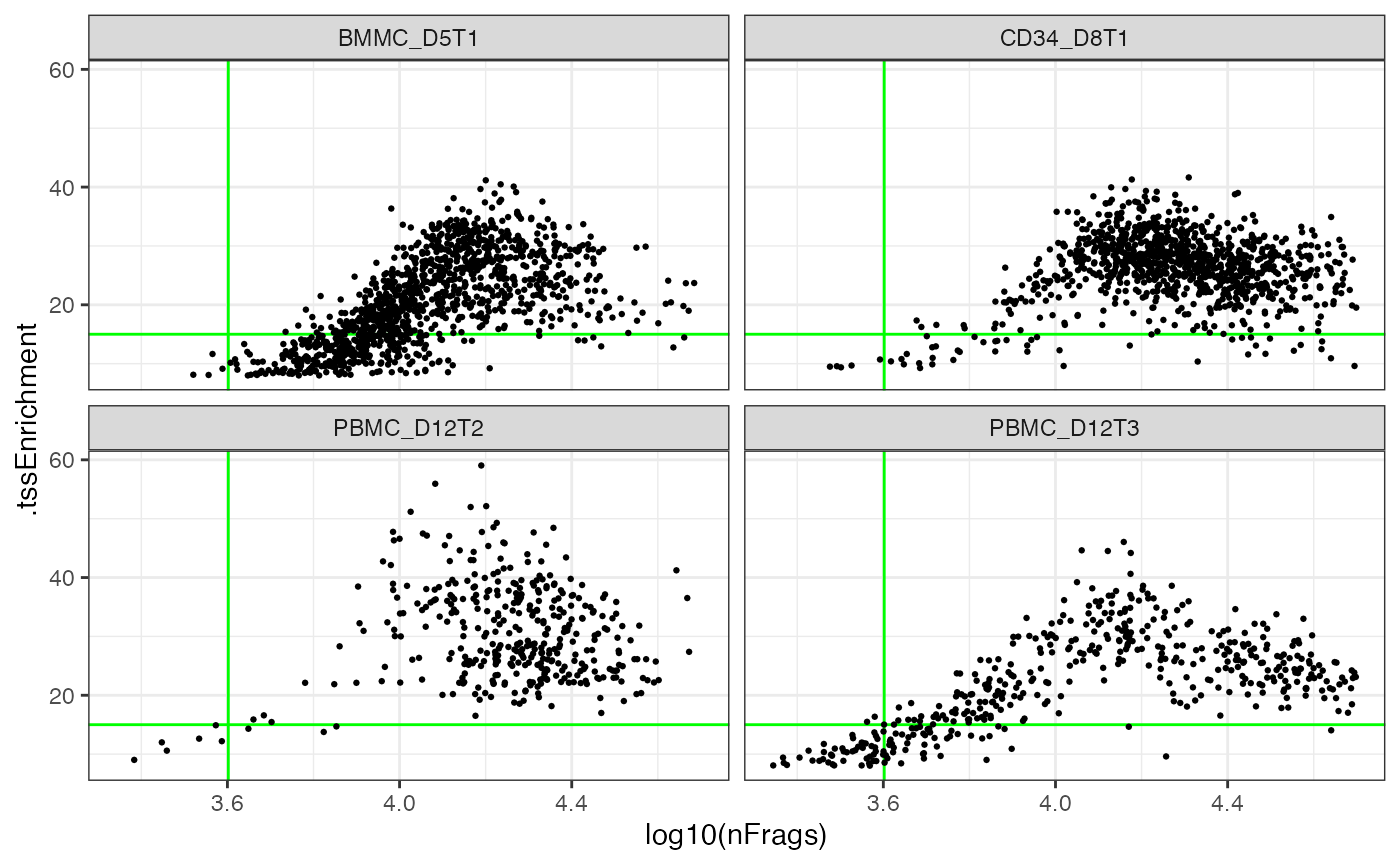
dsa_filtered <- dsa[cell_anno[,".tssEnrichment"] > cutoff_tss & cell_anno[,"nFrags"] > cutoff_nfrags]
length(getSamples(dsa))> [1] 3000
length(getSamples(dsa_filtered))> [1] 2527Exploratory analysis
Dimension reduction and clustering using iterative LSI
In order to obtain a low dimensional representation of single-cell ATAC datasets in terms of principal components and UMAP coordinates, we recommend an iterative application of the Latent Semantic Indexing approach (Cusanovich et al. 2018) described in (Granja et al. 2019). This approach also identifies cell clusters and a peak set that represents a consensus peak set of cluster peaks in a given dataset. In brief, in an initial iteration clusters are identified based on the most accessible regions (e.g. genomic tiling regions). Here, the counts are first normalized using the term frequency–inverse document frequency (TF-IDF) transformation and singular values are computed based on these normalized counts in selected regions (i.e. the most accessible regions in the initial iteration). Clusters are identified based on the singular values using Louvain clustering (as implemented in the Seurat package). Peak calling is then performed on the aggregated insertion sites from all cells of each cluster (using MACS2) and a union/consensus set of peaks uniform-length non-overlapping peaks is selected. In a second iteration, the peak regions whose TF-IDF-normalized counts which exhibit the most variability across the initial clusters provide the basis for a refined clustering using derived singular values. In the final iteration, the most variable peaks across the refined clusters are identified as the final peak set and singular values are computed again. Based on these final singular values UMAP coordinates are computed for low-dimensional projection.
# requires MACS2 for peak calling
itlsi <- iterativeLSI(dsa, it0regionType="tiling5kb", it0clusterResolution=0.4, it1clusterResolution=0.4, it2clusterResolution=0.4)
# load precomputed object
itlsi <- ChrAccRex::loadExample("itLsiObj_hema_example")The output object includes the final singular values/principal components (itlsi$pcaCoord), the low-dimensional coordinates (itlsi$umapCoord), the final cluster assignment of all cells (itlsi$clustAss), the complete, unfiltered initial cluster peak set (itlsi$clusterPeaks_unfiltered) as well as the final cluster-variable peak set (itlsi$regionGr):
str(itlsi$pcaCoord)> num [1:3000, 1:50] 0.0249 0.0242 0.023 0.0228 0.0232 ...
> - attr(*, "dimnames")=List of 2
> ..$ : chr [1:3000] "BMMC_D5T1_AAACTCGCAAATTGAG-1" "BMMC_D5T1_AAACTGCCACCAAGGA-1" "BMMC_D5T1_AAACTGCCAGTAAGAT-1" "BMMC_D5T1_AAACTGCCAGTCCTGG-1" ...
> ..$ : chr [1:50] "PC1" "PC2" "PC3" "PC4" ...
> - attr(*, "percVar")= num [1:50] 32.62 4.68 2.57 2.14 2.04 ...
> - attr(*, "PCAclass")= chr "PCcoord"
> - attr(*, "PCAmethod")= chr "irldba_svd"
> - attr(*, "SVD_D")= num [1:50, 1:50] 1.17 0 0 0 0 ...
> - attr(*, "SVD_U")= num [1:50000, 1:50] 0.00141 0.0013 0.00827 0.00411 0.0078 ...
> - attr(*, "SVD_V")= num [1:3000, 1:50] 0.0212 0.0206 0.0196 0.0194 0.0198 ...
str(itlsi$umapCoord)> num [1:3000, 1:2] -7.35 -7 -7.56 -5.6 -7.5 ...
> - attr(*, "dimnames")=List of 2
> ..$ : chr [1:3000] "BMMC_D5T1_AAACTCGCAAATTGAG-1" "BMMC_D5T1_AAACTGCCACCAAGGA-1" "BMMC_D5T1_AAACTGCCAGTAAGAT-1" "BMMC_D5T1_AAACTGCCAGTCCTGG-1" ...
> ..$ : chr [1:2] "UMAP1" "UMAP2"
str(itlsi$clustAss)> Factor w/ 7 levels "c0","c1","c2",..: 1 1 1 5 1 4 4 4 1 1 ...
> - attr(*, "names")= chr [1:3000] "BMMC_D5T1_AAACTCGCAAATTGAG-1" "BMMC_D5T1_AAACTGCCACCAAGGA-1" "BMMC_D5T1_AAACTGCCAGTAAGAT-1" "BMMC_D5T1_AAACTGCCAGTCCTGG-1" ...
length(itlsi$clusterPeaks_unfiltered)> [1] 300997
length(itlsi$regionGr)> [1] 50000You can use the resulting low-dimensional projections to characterize variability in your dataset according to various cell annotions. Here, we visualize the final cluster assignment …
df <- data.frame(
itlsi$umapCoord,
cluster = itlsi$clustAss,
cellId = rownames(itlsi$umapCoord),
sampleSource = getSampleAnnot(dsa)[,"cellType"],
stringsAsFactors = FALSE
)
ggplot(df, aes(x=UMAP1, y=UMAP2, color=cluster)) + geom_point(size=0.25)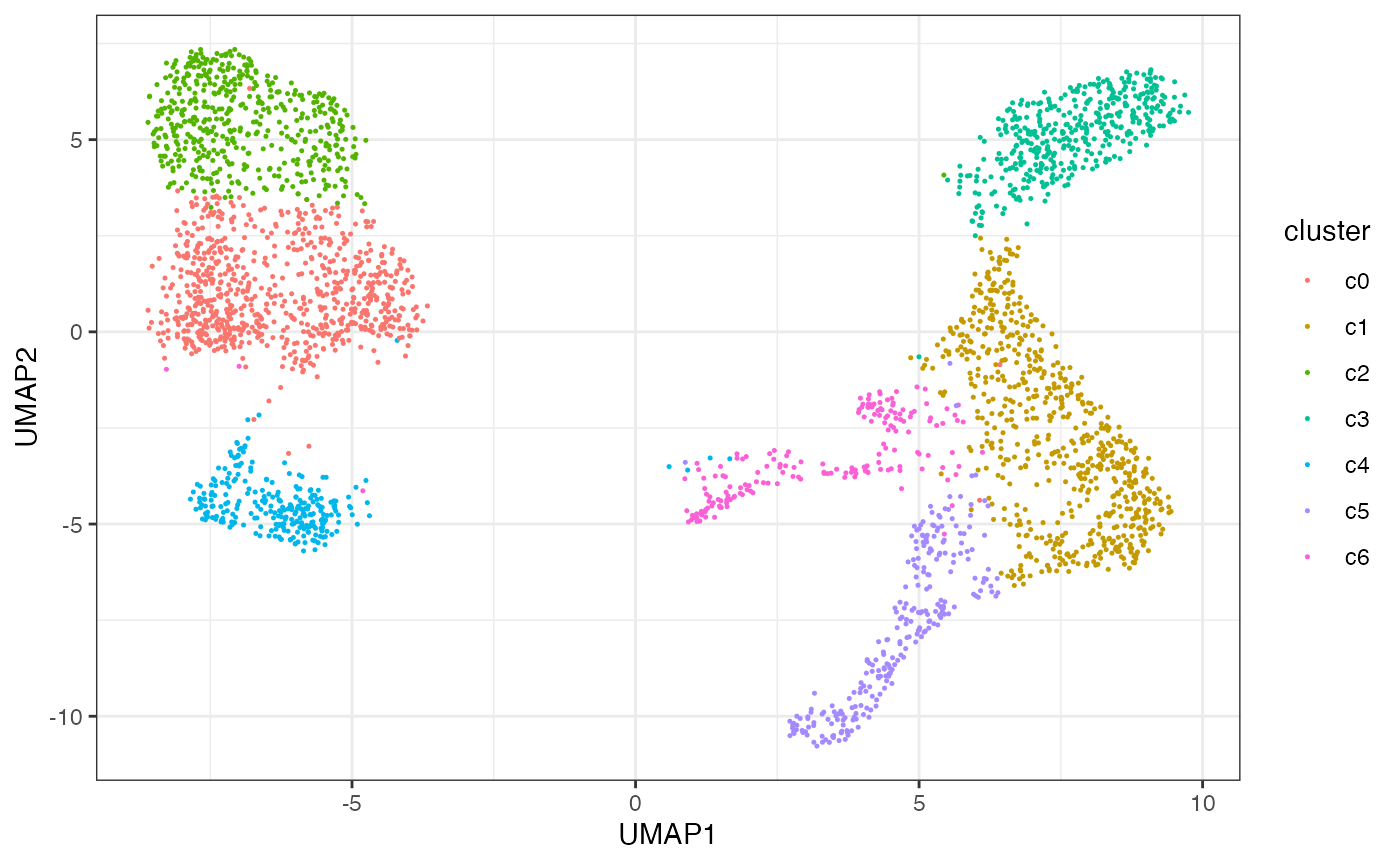
… as well as the annotated sample source:
ggplot(df, aes(x=UMAP1, y=UMAP2, color=sampleSource)) + geom_point(size=0.25)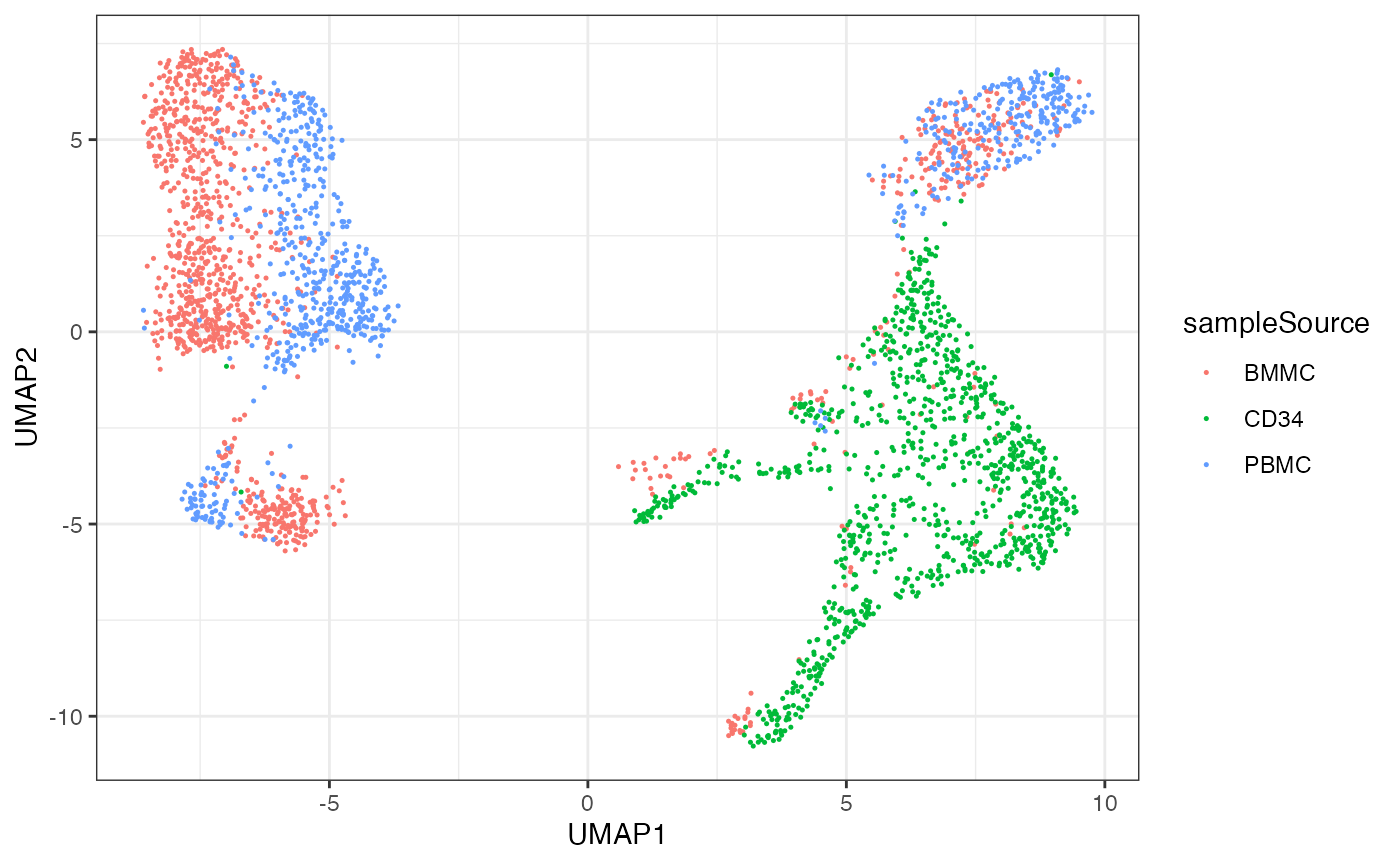
chromVAR deviations
We utilize chromVAR in order to determine the genome-wide TF motif activity for each cell (Schep et al. 2017). You can use the low-dimensional projection in order to visualize variability in TF motif accessibility across all cells in the manifold.
cvRes <- getChromVarDev(dsa, ".peaks.itlsi", motifs="jaspar")
devZ <- t(chromVAR::deviations(cvRes))
motifNames <- c("MA0036.3_GATA2", "MA0652.1_IRF8", "MA0102.3_CEBPA", "MA0690.1_TBX21")
df_with_cv <- data.frame(df, devZ[,motifNames])
plotL <- lapply(motifNames, FUN=function(mn){
ggplot(df_with_cv, aes_string(x="UMAP1", y="UMAP2", color=mn)) + geom_point(size=0.25) + scale_color_gradient2(midpoint=0, low="blue", mid="white", high="red")
})
do.call(cowplot::plot_grid, plotL)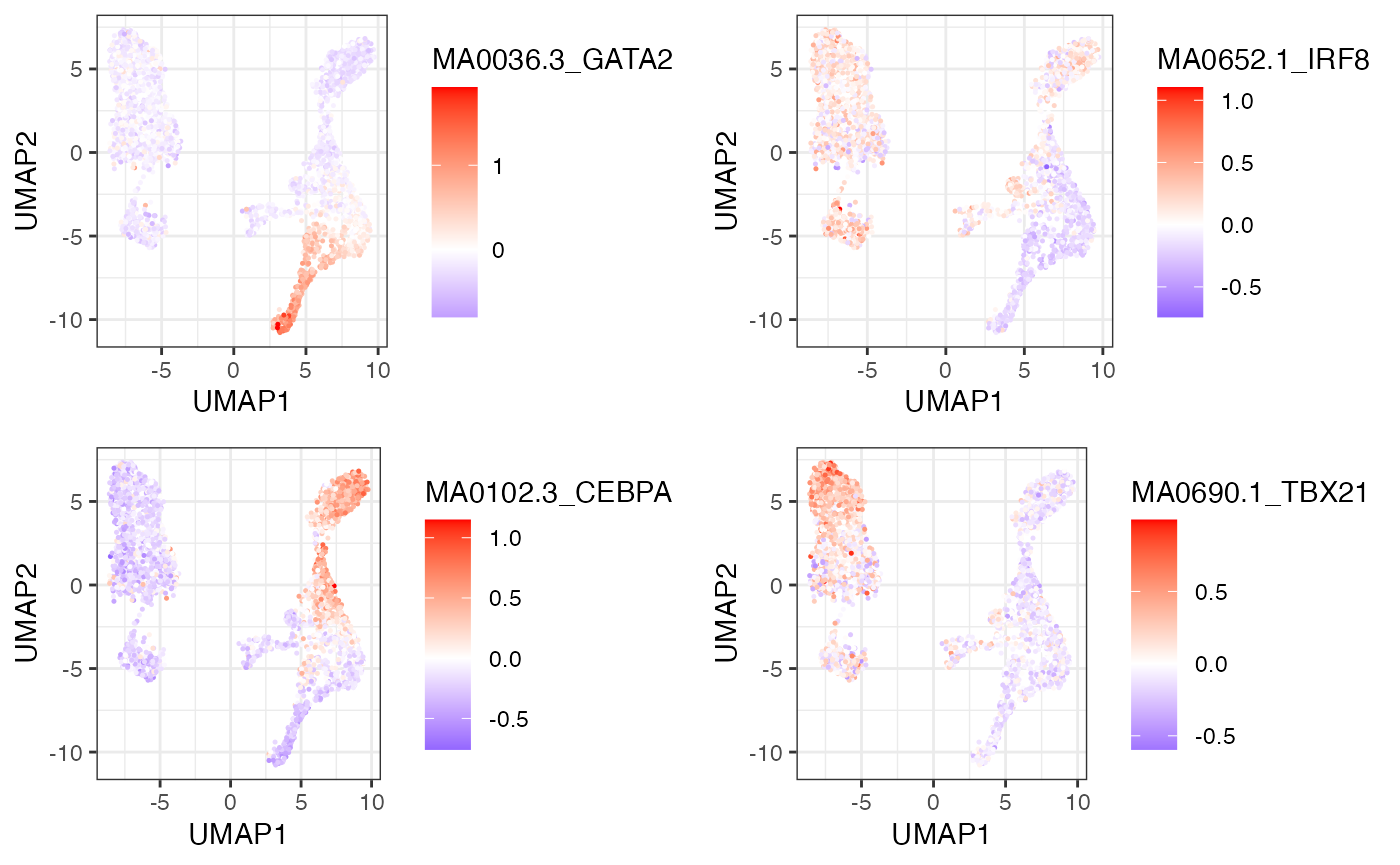
Gene activity
We can also define gene activity as the combined chromatin accessibility of a gene promoter and peaks that correlated with it across all accessibility profiles. This functionality is nicely implemented in the cicero and monocle packages and ChrAccR provides a wrapper function to compute gene activities from DsATAC datasets:
# requires monocle3 to be installed
library(monocle3)
promoter_gr <- getCoord(dsa, "promoter")
names(promoter_gr) <- GenomicRanges::elementMetadata(promoter_gr)[,"gene_name"]
gene_act <- getCiceroGeneActivities(
dsa,
".peaks.itlsi",
promoterGr=promoter_gr,
dimRedCoord=itlsi$pcaCoord
)Analyzing the full dataset
The full dataset of the single-cell human hematopoiesis study (Granja et al. 2019) is publicly available. Here, we show how ChrAccR can be applied to create a dataset from scratch using the aligned fragment files as input. Downstream steps can be run as shown above for the example subset of the data. Since the full dataset comprises high-quality profiles for a large number of cells, the analysis of such high-dimensional data has considerable resource requirements. We recommend to run it on a machine with a large amount of memory. The runtime for some of the folowing steps can exceed multiple hours.
Preparing the input
The main input for creating a single-cell dataset in ChrAccR are files containing the coordinates of sequenced fragments. Such fragment files can be obtained from the output of the CellRanger software (10x Genomics). For the hematopoiesis dataset analyzed here (Granja et al. 2019), these files are available from GEO (GEO accession: GSE139369)). First, download the GSE139369_RAW.tar file and unpack it (in the following, we assume that you unpacked the files to a directory called fragment_data). In addition to the fragment data, a sample annotation table that contains information on the samples’ phenotypes and technical parameters is needed. Here, we create one from the filenames:
fragmentFiles <- list.files("fragment_data", pattern=".fragments.tsv.gz")
sampleAnnotation <- data.frame(
sampleId = gsub("^(GSM[0-9]+)_scATAC_(.+).fragments\\.tsv\\.gz$", "\\2", fragmentFiles),
geoAccession = gsub("^(GSM[0-9]+)_scATAC_(.+).fragments\\.tsv\\.gz$", "\\1", fragmentFiles),
fragmentFilename = fragmentFiles,
stringsAsFactors = FALSE
)
sampleAnnotation[,"source"] <- gsub("^(MPAL|CD34|BMMC|PBMC).+", "\\1", sampleAnnotation[,"sampleId"])
# only samples from healthy donors
sampleAnnotation <- sampleAnnotation[sampleAnnotation[,"source"]!="MPAL",]
inputFiles <- file.path('fragment_data', sampleAnnotation[,"fragmentFilename"])Vanilla analysis
As demonstrated in the overview vignette, ChrAccR can run a default analysis workflow by using the run_atac function. For single-cell datasets, this requires you to supply a reference to the fragment files or the output of the CellRanger software, as well a sample annotation table:
setConfigElement("annotationColumns", c(".sampleId", "cellType", "nFrags", "tssEnrichment"))
dsa_full <- run_atac(config$.anaDir, input=inputFiles, sampleAnnot=sampleAnnotation, genome="hg19", sampleIdCol="sampleId")Analysis steps in detail
Preparing the DsATAC data structure from fragment files
Then we can directly create a DsATACsc dataset from the fragment files:
dsa_full_raw <- DsATACsc.fragments(
sampleAnnotation,
inputFiles,
"hg19",
regionSets=NULL,
sampleIdCol="sampleId",
minFragsPerBarcode=1000L,
maxFragsPerBarcode=50000L,
keepInsertionInfo=TRUE
)Filtering low quality cells
It is recommended to filter low quality cells from the datasets. In the code above, we already set a threshold for the minimum and maximum number of fragments for each cell in order for it to be included in the dataset. The following code also limits the dataset to cells that exhibit a sufficiently high signal-to-noise ratio, as defined by the enrichment of Tn5 insertions at transcription start sites (TSS) relative to background.
tsse_cutoff <- 8
dsa_full <- filterCellsTssEnrichment(dsa_full_raw, tsse_cutoff)Dimension reduction and clustering using iterative LSI
We recommend to use the iterative LSI approach outlined above to obtain a relatively robust dimensionality reduction and clustering of cells. We compute the reduced components and coordinates in the code below. We also add the assigned cluster for each cell to the dataset annotation and aggregate the Tn5 insertion counts for the computed consensus peak set.
itlsi <- iterativeLSI(dsa_full, it0regionType="tiling5kb", it0clusterResolution=0.4, it1clusterResolution=0.4, it2clusterResolution=0.4)
# add cluster assignment to cell annotation
dsa_full <- addSampleAnnot(dsa_full, "cluster_itlsi", itlsi$clustAss)
# aggregate insertions accross the consensus set of cluster peaks derived during iterative LSI
dsa_full <- regionAggregation(dsa_full, itlsi$clusterPeaks_unfiltered, "peaks_itlsi", signal="insertions", dropEmpty=FALSE, bySample=FALSE)Analysis reports
ChrAccR also provides methods to automatically generate analysis reports that summarize most of the above analysis steps. The reports for the full single-cell dataset described here can also be found in the ‘Resource’ section of the ChrAccR website. Provided with a DsATAC dataset these reports can be generated with a call to the corresponding createReport_* function. For instance, the createReport_summary generates a report containing an overview of sample and cell statistics:
createReport_summary(dsa_full, file.path("reports"))The report generated by the createReport_exploratory functions comprises unsupervised analysis, including dimensionality reduction and the quantification of transcription factor activities. Analysis parameters for generating these reports can be configures using the setConfigElement function.
chromVarMotifsToPlot <- c("MA0036.3_GATA2", "MA0652.1_IRF8", "MA0102.3_CEBPA", "MA0466.2_CEBPB", "MA1141.1_FOS::JUND", "MA0080.4_SPI1", "MA0105.4_NFKB1", "MA0014.3_PAX5", "MA0002.2_RUNX1", "MA0690.1_TBX21")
setConfigElement("annotationColumns", c(".sampleId", "cellType", "nFrags", "tssEnrichment"))
setConfigElement("scIterativeLsiRegType", "tiling5kb")
setConfigElement("scIterativeLsiClusterResolution", 0.4)
setConfigElement("chromVarMotifNamesForDimRed", chromVarMotifsToPlot)
createReport_exploratory(dsa_full, file.path("reports"))Session Info
Sys.Date()> [1] "2023-03-17"> R version 4.2.2 (2022-10-31)
> Platform: aarch64-apple-darwin20 (64-bit)
> Running under: macOS Ventura 13.2.1
>
> Matrix products: default
> BLAS: /Library/Frameworks/R.framework/Versions/4.2-arm64/Resources/lib/libRblas.0.dylib
> LAPACK: /Library/Frameworks/R.framework/Versions/4.2-arm64/Resources/lib/libRlapack.dylib
>
> locale:
> [1] C/UTF-8/C/C/C/C
>
> attached base packages:
> [1] stats graphics grDevices utils datasets methods base
>
> other attached packages:
> [1] ggplot2_3.4.1 ChrAccR_0.9.21
>
> loaded via a namespace (and not attached):
> [1] colorspace_2.1-0 rjson_0.2.21
> [3] ellipsis_0.3.2 rprojroot_2.0.3
> [5] XVector_0.38.0 GenomicRanges_1.50.2
> [7] fs_1.6.1 farver_2.1.1
> [9] DT_0.27 bit64_4.0.5
> [11] AnnotationDbi_1.60.2 fansi_1.0.4
> [13] R.methodsS3_1.8.2 codetools_0.2-19
> [15] cachem_1.0.7 knitr_1.42
> [17] jsonlite_1.8.4 Rsamtools_2.14.0
> [19] seqLogo_1.64.0 annotate_1.76.0
> [21] GO.db_3.16.0 R.oo_1.25.0
> [23] png_0.1-8 shiny_1.7.4
> [25] httr_1.4.5 readr_2.1.4
> [27] compiler_4.2.2 lazyeval_0.2.2
> [29] ChrAccRex_0.2 Matrix_1.5-3
> [31] fastmap_1.1.1 cli_3.6.0
> [33] later_1.3.0 muLogR_0.2
> [35] htmltools_0.5.4 tools_4.2.2
> [37] gtable_0.3.1 glue_1.6.2
> [39] TFMPvalue_0.0.9 GenomeInfoDbData_1.2.9
> [41] reshape2_1.4.4 dplyr_1.1.0
> [43] Rcpp_1.0.10 Biobase_2.58.0
> [45] jquerylib_0.1.4 pkgdown_2.0.7
> [47] vctrs_0.6.0 Biostrings_2.66.0
> [49] rtracklayer_1.58.0 xfun_0.37
> [51] CNEr_1.34.0 stringr_1.5.0
> [53] mime_0.12 miniUI_0.1.1.1
> [55] lifecycle_1.0.3 restfulr_0.0.15
> [57] poweRlaw_0.70.6 gtools_3.9.4
> [59] XML_3.99-0.13 zlibbioc_1.44.0
> [61] scales_1.2.1 JASPAR2018_1.1.1
> [63] BSgenome_1.66.3 ragg_1.2.5
> [65] hms_1.1.2 promises_1.2.0.1
> [67] MatrixGenerics_1.10.0 parallel_4.2.2
> [69] SummarizedExperiment_1.28.0 yaml_2.3.7
> [71] memoise_2.0.1 chromVAR_1.20.2
> [73] sass_0.4.5 stringi_1.7.12
> [75] RSQLite_2.3.0 highr_0.10
> [77] S4Vectors_0.36.2 BiocIO_1.8.0
> [79] desc_1.4.2 caTools_1.18.2
> [81] BiocGenerics_0.44.0 BiocParallel_1.32.5
> [83] GenomeInfoDb_1.34.9 rlang_1.1.0
> [85] pkgconfig_2.0.3 systemfonts_1.0.4
> [87] matrixStats_0.63.0 bitops_1.0-7
> [89] pracma_2.4.2 evaluate_0.20
> [91] lattice_0.20-45 purrr_1.0.1
> [93] GenomicAlignments_1.34.1 htmlwidgets_1.6.1
> [95] labeling_0.4.2 cowplot_1.1.1
> [97] bit_4.0.5 tidyselect_1.2.0
> [99] BSgenome.Hsapiens.UCSC.hg19_1.4.3 RcppAnnoy_0.0.20
> [101] plyr_1.8.8 magrittr_2.0.3
> [103] R6_2.5.1 IRanges_2.32.0
> [105] generics_0.1.3 DelayedArray_0.24.0
> [107] DBI_1.1.3 pillar_1.8.1
> [109] withr_2.5.0 KEGGREST_1.38.0
> [111] RCurl_1.98-1.10 tibble_3.2.0
> [113] crayon_1.5.2 muRtools_0.9.5
> [115] utf8_1.2.3 plotly_4.10.1
> [117] nabor_0.5.0 tzdb_0.3.0
> [119] rmarkdown_2.20 TFBSTools_1.36.0
> [121] grid_4.2.2 data.table_1.14.8
> [123] blob_1.2.3 digest_0.6.31
> [125] xtable_1.8-4 tidyr_1.3.0
> [127] httpuv_1.6.9 R.utils_2.12.2
> [129] textshaping_0.3.6 stats4_4.2.2
> [131] munsell_0.5.0 DirichletMultinomial_1.40.0
> [133] viridisLite_0.4.1 motifmatchr_1.20.0
> [135] bslib_0.4.2